
Sisältö
Kystinen fibroosi (CF) on perinnöllinen, hengenvaarallinen häiriö, joka vahingoittaa keuhkoja ja ruoansulatuskanavaa. Sen aiheuttaa viallinen geeni, joka laukaisee sakeutuneen liman tuotannon, joka tukkii hengitystiet ja estää ruoansulatusentsyymien erityksen.Oireet ovat progressiivisia ja usein vakavia, ja niihin voi sisältyä hengitysvaikeuksia, toistuvia keuhkoinfektioita, heikkoa kasvua, miesten hedelmättömyyttä ja kroonista haiman, maksan, munuaisten ja sydämen tulehdusta.
CF voidaan diagnosoida verikokeilla, geneettisellä seulonnalla ja higikloriditestinä tunnetulla menettelyllä.
Vaikka CF: lle ei ole parannuskeinoa, on hoitoja, jotka voivat parantaa sekä elämän pituutta että laatua.
Näitä ovat hengitysteiden puhdistumistekniikat, hengitettävät antibiootit, limanohennusaineet, haimaentsyymit, runsaskalorinen ruokavalio ja uuden sukupolven lääkkeet, jotka tunnetaan nimellä CFTR-modulaattorit. Vaikeissa tapauksissa keuhkosiirto voi olla tarpeen.
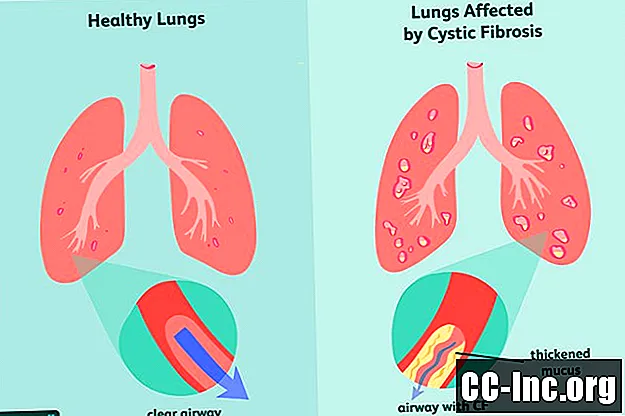
Kystisen fibroosin oireet
Geneettisenä häiriönä kystinen fibroosi on jotain, jolla olet syntynyt. Sillä voi olla oireita syntymähetkellä tai ei, ja se voi usein kestää kuukausia tai jopa vuosia ennen kuin oireita ilmenee. Siihen mennessä keuhkot ja ruoansulatuskanava ovat jo saattaneet kokea vaurioita, joita ei voida kumota.
CF: n yleisimpiä varhaisia oireita ovat:
- Vauvan ensimmäisen jakkara (mekonium) tukkeutuu
- Suolamainen maku
- Krooninen yskä, hengityksen vinkuminen tai värillinen yskökset
- Löysä, rasvainen ja tyypillisesti hajuinen uloste
- Keuhkoinfektio, usein toistuva
- Huono kasvu ja menestymisen epäonnistuminen
Ellei näitä oireita voida hallita, keuhkoilla esiintyvällä stressillä (ja kyvyttömyydellä painonnousuun) voi olla kumulatiivinen vaikutus, joka vaikuttaa useisiin elimiin ja lisää taudin komplikaatioiden riskiä.
Joitakin tyypillisempiä komplikaatioita ovat:
- Viivästynyt murrosikä
- Bronkiektaasi (keuhkoseinämien krooninen paksuuntuminen)
- Painonpudotus
- Haimatulehdus (haimatulehdus)
- Miehen hedelmättömyys
- Keuhkoverenpainetauti (korkea verenpaine keuhkoissa)
- Sappikivet
- Kystiseen fibroosiin liittyvä diabetes
- Cor pulmonale (oikeanpuoleinen sydämen vajaatoiminta)
- Kirroosi (maksan toiminnallinen arpeutuminen)
Koska CF aiheuttaa progressiivista vahinkoa soluille ja kudoksille, kaikki keuhkoihin ja muihin elimiin aiheutuvat vauriot ovat suurelta osin peruuttamattomia. Kuolema johtuu useimmiten hengitysvajauksesta, jota seuraa sydämen vajaatoiminta ja maksan vajaatoiminta.
Kystisen fibroosin oireet
Syyt
Kystinen fibroosi johtuu CFTR-proteiinin tuottamisesta vastaavan kystisen fibroosin transmembraanireseptorin (CFTR) geenin mutaatiosta.Tämä on proteiini, jota keho tarvitsee säätelemään suolan ja veden virtausta soluissa ja soluista . Jos proteiini on epämuodostunut tai viallinen, se voi aiheuttaa dehydraation solun pinnalla, mikä johtaa ympäröivän liman sakeutumiseen.
CF on autosomaalinen resessiivinen häiriö, mikä tarkoittaa, että sinun on perittävä CFTR-mutaatio sekä äidiltäsi että isältäsi sairauden saamiseksi. Jos perit vain yhden viallisen geenin, sinulla ei ole CF: tä, vaan olet sen sijaan mutatoidun geenin kantaja.
Voit periä taudin, jos jokaisella vanhemmastasi on joko CFTR-mutaatio tai itse CF. Jos molemmat vanhemmat ovat kuljettajia, sinulla on:
- 25 prosentin mahdollisuus saada CF
- 50 prosentin mahdollisuus olla lentoliikenteen harjoittaja
- 25 prosentin mahdollisuus tulla ennalleen
Toisaalta, jos toinen vanhemmistasi on kantaja ja toisella on CF, sinulla on 50/50 mahdollisuus joko saada CF tai olla kantaja.
Kystinen fibroosi on yksi yleisimmistä geneettisistä sairauksista, ja se vaikuttaa noin jokaiseen 2500: sta Yhdysvalloissa syntyneestä vauvasta.
Se on yleisintä valkoihoisten ja latinalaisamerikkalaisten keskuudessa, ja sitä esiintyy harvemmin afrikkalaisista tai aasialaisista ihmisistä.
Kystisen fibroosin riskitekijätDiagnoosi
Kystisen fibroosin diagnosoinnissa käytetään muutamia testejä. Ne toimivat joko havaitsemalla suoraan CFTR-mutaatio tai mittaamalla epäsuorasti taudin mukaisia biologisia muutoksia. Diagnoosimenetelmä voi vaihdella raskauden aikana, vauvan syntyessä tai milloin tahansa sen jälkeen.
Kystisen fibroosin lääkärin keskusteluopas
Hanki tulostettava oppaamme seuraavaa lääkärisi tapaamista varten, jotta voit kysyä oikeita kysymyksiä.

Kahdesta vakiotestistä, joita yleisesti käytetään CF: n diagnosointiin:
- Hikikloridin testaus, joka tunnetaan myös yksinkertaisesti hiki-testinä, mittaa kloridin määrää iholla. Koska CF häiritsee suolan siirtymistä soluihin ja soluista, suolaan kertyy hikeä.
- Geneettinen CFTR-testaus käytetään havaitsemaan CFTR-mutaation yleisimmät mutaatiot. Vaikka kystistä fibroosia tiedetään aiheuttavan yli 2000 CFTR-mutaatiota, standardipaneeliin sisältyvät 23 edustavat todennäköisimpiä epäiltyjä.
Raskauden aikana CFTR-geneettistä testiä voidaan käyttää lapsivesitutkimuksen avulla saatujen nesteiden tai korionvillanäytteenoton (CVS) avulla uutettujen solujen testaamiseen.
Vastasyntyneen seulonta Sitä käytetään myös vakavasti CF: n diagnosoinnissa, ja se on nykyään valtuutettu kaikissa 50 osavaltiossa ja Columbian piirikunnassa. Jos vastasyntyneen seulontatulokset ovat positiivisia, diagnoosin vahvistamiseksi käytetään hiki-testiä.
Kuinka kystinen fibroosi diagnosoidaanHoito
Vaikka kystiseen fibroosiin ei ole parannuskeinoa, hoidon edistyminen on pidentänyt taudissa elävien ikää.
CF-hoidon tavoitteena on nelinkertainen: estää infektioita, ylläpitää keuhkotoimintaa, normalisoida ruoansulatusta ja hidastaa taudin etenemistä.
CF-hoidossa käytettyjen terapeuttisten välineiden joukossa:
- Hengitysteiden puhdistustekniikat (ACT) suoritetaan kertyneen liman poistamiseksi ja poistamiseksi keuhkoista. Tekniikoita ovat huff yskä, rintakehän lyömäsoittimet tai rintaseinän värähtely.
- Rasvainen ja kaloripitoinen ruokavalio käytetään kompensoimaan suolistossa olevien rasvojen, proteiinien ja ravintoaineiden imeytymishäiriö.
- Haiman entsyymilisät käytetään vahvistamaan ruuansulatusentsyymejä, joita haima ei voi tuottaa liman liiallisen kertymisen vuoksi.
- Antibiootit otetaan päivittäin bakteeri-infektioiden estämiseksi.
- Mukolyytit- lääkkeitä, joita käytetään liman ohentamiseen ennen ACT: ita, voidaan käyttää.
- CFTR-modulaattorit ovat uusi lääkeryhmä, joka voi korjata tietyt CFTR-proteiinin puutteet ja palauttaa niiden säätelytoiminnon.
- Happihoito Sitä voidaan käyttää akuuttien jaksojen aikana, kun hengitys on vakavasti heikentynyt.
- Enteraalinen ravitsemus, tunnetaan myös nimellä putkisyöttö, voidaan käyttää, jos et voi ylläpitää painoa normaalilla ravinnolla.
- Keuhkojensiirto pidetään silloin, kun keuhkot eivät enää tue selviytymistä ilman mekaanista tuuletusta.
Selviytyminen
Vuonna 1938, kun kystinen fibroosi luokiteltiin ensimmäisen kerran sairaudeksi, lapset elivät harvoin ensimmäisen elinvuodensa jälkeen. 1980-luvulle mennessä voidaan odottaa elävänsä jopa 20-25 vuotta. Nykyään kuva on muuttunut kokonaan ihmisten kanssa, jotka elävät hyvin 40- ja jopa 50-vuotiaina, jos hoito aloitetaan aikaisin ja noudatetaan.
Tämä ei tarkoita sitä, että CF olisi yhtä vakava kuin koskaan. Se on elämää muuttava tapahtuma, joka vaatii huolellisuutta ja johdonmukaisuutta paitsi selviytyä taudista myös elää korkeinta mahdollista elintasoa.
Tätä varten sinun on normalisoitava CF elämässäsi asettamalla rutiinit ja käytännöt, jotta vältetään ylä- ja alamäet, jotka voivat aiheuttaa stressiä ja lisätä vammaisuutta. Huomioiden joukossa sinun on:
- Hallitse ravintoa. CF-potilaat tarvitsevat usein kaksi kertaa päivässä enemmän kaloreita kuin muut ihmiset.
- Harjoittele säännöllisesti. Kuntorutiinien tulisi ihannetapauksessa sisältää vähintään 20-30 minuuttia aerobista toimintaa kolme kertaa viikossa. Löydä jotain nautittavaa, jonka voit tehdä koko eliniän.
- Pidä hyvin hydratoituna. Näin pitämällä keuhkot ja suolet toimivat kunnolla. Iästä riippuen sinun tulisi juoda vähintään kuusi - kahdeksan korkeaa lasillista vettä päivässä.
- Suorita hengitysteiden puhdistuma oikein. Kun terveystarpeesi muuttuvat, myös tarvitsemasi puhdistustyökalut voivat muuttua. Keskustele pulmonologin tai fysioterapeutin kanssa, jos et saavuta tuloksia, joita sinun pitäisi.
- Etsi tukea. Ystävien ja perheen lisäksi voit ottaa yhteyttä kystisen fibroosin säätiön (CFF) lähimpään osastoon liittääksesi tukiverkkoon alueellasi.
- Pyydä taloudellista apua. CFF tarjoaa palveluja, jotka auttavat perheitä selviytymään paremmin CF-hoidon korkeista kustannuksista.
Sana Verywelliltä
Vaikka vastasyntyneiden seulonnat ovat lisänneet dramaattisesti vauvojen CF-diagnoosien määrää, yli 25 prosenttia diagnooseista tehdään vain lapsuudessa, teini-ikäisillä ja varhaisilla aikuisilla.
Tämä on ongelmallista, koska varhainen diagnoosi ja hoito voivat estää monia CF: n vakavampia komplikaatioita ennen kuin vakavia vaurioita voidaan tehdä. Hoito ei pysty pysäyttämään tai kääntämään tautia, mutta se voi taata monia muita tauteettomia vuosia.
Tätä varten on tärkeää tietää CF: n varhaiset oireet ja keskustella lääkärisi kanssa, jos epäilet lapsellasi olevan sairaus. Tämä pätee erityisesti valtioihin, joissa näytöllä käytetään vain IRT-verikokeita, mikä voi johtaa siihen, että jopa 5 prosentilla lapsista on joko viivästynyt diagnoosi tai väärän negatiivinen tulos, Wisconsinin yliopiston lääketieteen ja kansanterveyden korkeakoulun tutkimuksen mukaan .
Mitä oireita voit odottaa kystisen fibroosin yhteydessä?