
Sisältö
- syyt
- oireet
- Tentit ja testit
- hoito
- Tukiryhmät
- Outlook (ennuste)
- Kun otat yhteyttä lääkäriin
- ennaltaehkäisy
- Vaihtoehtoiset nimet
- kuvat
- Viitteet
- Arvostelun päivämäärä 10/15/2018
Niemann-Pick-tauti (NPD) on joukko sairauksia, jotka kulkevat perheet (perinnölliset), joissa rasva-aineet, joita kutsutaan lipideiksi, kerääntyvät pernan, maksan ja aivojen soluihin.
Taudin yleisiä muotoja on kolme:
- A tyypin
- Tyyppi B
- Tyyppi C
Jokainen tyyppi sisältää erilaisia elimiä. Se voi tai ei koske hermostoa ja hengitystä. Jokainen voi aiheuttaa erilaisia oireita ja saattaa esiintyä eri aikoina koko elämän ajan.
syyt
NPD-tyypit A ja B ilmenevät, kun elimistössä olevilla soluilla ei ole entsyymiä, jota kutsutaan happosfingomyelinaasiksi (ASM). Tämä aine auttaa hajottamaan (metaboloitumaan) sfingomyeliini-nimisen rasva-aineen, joka löytyy jokaisesta kehon solusta.
Jos ASM puuttuu tai se ei toimi kunnolla, sfingomyeliini kerääntyy solujen sisällä. Tämä tappaa solusi ja vaikeuttaa elinten toimintaa.
Tyyppi A esiintyy kaikissa rotuissa ja etnisissä ryhmissä. Se on yleisempää Ashkenazin (Itä-Euroopan) juutalaisväestössä.
Tyyppi C tapahtuu, kun elimistö ei pysty hajottamaan kolesterolia ja muita rasvoja (lipidejä). Tämä johtaa liian suureen kolesteroliarvoon maksassa ja pernassa ja liian suuressa määrin muita lipidejä aivoissa. Tyyppi C on yleisin espanjalaisen laskujen Puerto Ricansissa.
Tyyppi C1 on tyypin C muunnos. Se sisältää vian, joka häiritsee kolesterolin siirtymistä aivosolujen välillä. Tätä tyyppiä on nähty vain Ranskan kanadalaisissa ihmisissä Yarmouthin piirikunnassa Nova Scotiassa.
oireet
Oireet vaihtelevat. Muut terveysolosuhteet voivat aiheuttaa samanlaisia oireita. Taudin alkuvaiheet voivat aiheuttaa vain muutamia oireita. Henkilöllä ei ehkä ole mitään oireita.
Tyyppi A alkaa yleensä ensimmäisten elämänkuukausien aikana. Oireita voivat olla:
- Vatsan turvotus 3–6 kuukauden kuluessa
- Kirsikanpunainen piste silmän takana (verkkokalvolla)
- Ruokintaongelmat
- Varhaisen moottoritaidon menetys (pahenee ajan myötä)
Tyypin B oireet ovat yleensä lievempiä. Ne esiintyvät myöhäisessä lapsuudessa tai teini-ikäisinä. Pienillä lapsilla voi esiintyä vatsan turvotusta. Aivoja ja hermostoa ei ole lainkaan osallisena, kuten motoristen taitojen menetys. Joillakin lapsilla voi olla toistuvia hengitystieinfektioita.
Tyypit C ja C1 vaikuttavat yleensä kouluikäisiin lapsiin. Se voi kuitenkin esiintyä milloin tahansa varhaislapsuuden ja aikuisen välillä. Oireita voivat olla:
- Raajojen liikkuvuus, joka voi johtaa epävakaan kulkuun, kömpelykseen, kävelyongelmiin
- Laajennettu perna
- Laajennettu maksa
- Keltaisuus syntymässä (tai pian sen jälkeen)
- Oppimisvaikeudet ja henkinen lasku
- takavarikot
- Epäsäännöllinen, epäsäännöllinen puhe
- Äkillinen lihasten äänen menetys, joka voi johtaa putoamiseen
- vapina
- Ongelma siirtää silmät ylös ja alas
Tentit ja testit
Veren tai luuytimen testi voidaan tehdä tyypin A ja B diagnosoimiseksi. Testi voi kertoa, kenellä on tauti, mutta ei näytä, olisitko kantaja. DNA-testit voidaan tehdä tyypin A ja B kantajien diagnosoimiseksi.
Ihon biopsia tehdään yleensä tyypin C ja D diagnosoimiseksi. Terveydenhuollon tarjoaja valvoo, miten ihosolut kasvavat, liikkuvat ja säilyttävät kolesterolia. DNA-testejä voidaan myös tehdä kahden geenin etsimiseksi, jotka aiheuttavat tämäntyyppisen taudin.
Muita testejä voivat olla:
- Luuytimen pyrkimys
- Maksabiopsia (yleensä ei tarvita)
- Slit-lampun silmätutkimus
- Testit ASM-tason tarkistamiseksi
hoito
Tällä hetkellä ei ole olemassa tehokasta hoitoa tyypille A.
Luunydinsiirtoja voidaan kokeilla tyypin B osalta. Tutkijat tutkivat edelleen mahdollisia hoitoja, mukaan lukien entsyymin korvaaminen ja geeniterapia.
Uusi lääke, jota kutsutaan miglustaatiksi, on saatavilla tyypin C hermoston oireille.
Korkeaa kolesterolia voidaan hoitaa terveellä, alhaisella kolesterolia sisältävällä ravinnolla tai lääkkeillä. Tutkimukset eivät kuitenkaan osoita, että nämä menetelmät estävät taudin pahenemisen tai muuttavat solujen kolesterolin hajoamista. Lääkkeitä on saatavilla monien oireiden hallitsemiseksi tai lievittämiseksi, kuten äkillinen lihasten värjäys ja kohtaukset.
Tukiryhmät
Nämä järjestöt voivat tarjota tukea ja lisätietoja Niemann-Pick-taudista:
- Kansallinen neurologisten häiriöiden ja aivohalvauksen laitos - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Kansallinen Niemann-Pick-taudin säätiö - nnpdf.org
- Harvinaisia sairauksia käsittelevä kansallinen järjestö - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Outlook (ennuste)
NPD-tyyppi A on vakava sairaus. Se johtaa yleensä kuolemaan 2 tai 3-vuotiaana.
Ne, joilla on tyyppi B, voivat elää myöhään lapsuudessa tai aikuisuudessa.
Lapsi, joka näyttää tyypin C merkkejä ennen 1-vuotiaita, ei ehkä asu kouluikäiseksi. Ne, jotka osoittavat oireita koulunkäynnin jälkeen, voivat elää keski-ikäisillään. Jotkut voivat elää 20-luvulla.
Kun otat yhteyttä lääkäriin
Tee tapaaminen palveluntarjoajan kanssa, jos sinulla on perheen historiaa Niemann-Pick-taudissa ja aiot saada lapsia. Geneettinen neuvonta ja seulonta on suositeltavaa.
Soita palveluntarjoajalle, jos lapsellasi on taudin oireita, mukaan lukien:
- Kehitysongelmat
- Syöttöongelmat
- Huono painonnousu
ennaltaehkäisy
Kaikki Niemann-Pickin tyypit ovat autosomaalisia resessiivisiä. Tämä tarkoittaa, että molemmat vanhemmat ovat kuljettajia. Jokaisella vanhemmalla on 1 kopio epänormaalista geenistä ilman mitään merkkejä taudista itsestään.
Kun molemmat vanhemmat ovat liikenteenharjoittajia, on 25 prosentin todennäköisyys, että heidän lapsensa sairastavat taudin ja 50 prosentin todennäköisyys, että heidän lapsensa on kantaja.
Kantoaallon tunnistustestaus on mahdollista vain, jos geneettinen vika on tunnistettu. Tyyppeihin A ja B liittyvät viat on tutkittu hyvin. Näiden Niemann-Pick-muotojen DNA-testit ovat saatavilla.
Monien C-tyypin DNA: ssa on havaittu geneettisiä vikoja. Voi olla mahdollista diagnosoida epänormaalia geeniä kantavia ihmisiä.
Muutamat keskukset tarjoavat testejä vauvan diagnosoimiseksi vielä kohdussa.
Vaihtoehtoiset nimet
NPD; Sfingomyelinaasin puutos; Lipidien varastointihäiriö - Niemann-Pickin tauti; Lysosomaalisen varastoinnin tauti - Niemann-Pick
kuvat
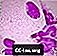
Niemann-Pick vaahdotetut solut
Viitteet
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Lipidien metabolian puutteet. Julkaisussa: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, toim. Nelsonin lastenlääketieteen oppikirja. 20th ed. Philadelphia, PA: Elsevier; 2016: kappale 86.
Patterson MC, Hendriksz CJ; NP-C-suuntaviivojen työryhmä et al. Suositukset C-tyypin Niemann-Pick-taudin diagnosointiin ja hoitoon: päivitys. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Metabolian synnynnäiset virheet. Julkaisussa: Turnpenny PD, Ellard S, toim. Emeryn lääketieteellisen genetiikan elementit. 15th ed. Philadelphia, PA: Elsevier; 2017: luku 18.
Arvostelun päivämäärä 10/15/2018
Päivitetty: Anna C. Edens Hurst, MD, MS, lääketieteellisen genetiikan apulaisprofessori, Alabaman yliopisto Birminghamissa, Birminghamissa, AL. VeriMed Healthcare Networkin toimittama arvostelu. Tarkastellut myös David Zieve, MD, MHA, lääketieteellinen johtaja, toimittajajohtaja Brenda Conaway ja A.D.A.M. Toimituksellinen tiimi.